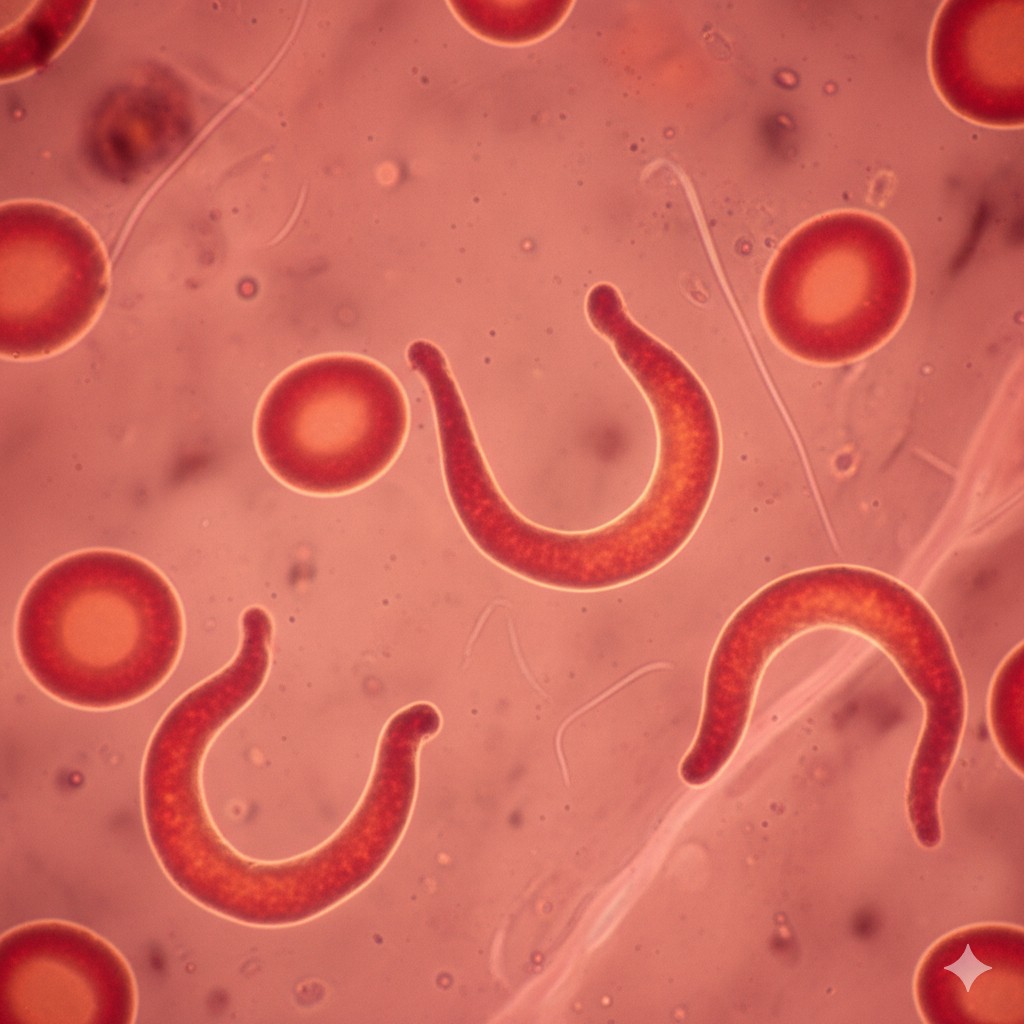
Sickle cell disease (SCD) was once described primarily as a hereditary anemia caused by an abnormal hemoglobin molecule1. In this condition, a mutation in the β-globin (HBB) gene produces hemoglobin S (HbS), a variant that causes red blood cells to adopt a rigid, crescent or “sickle” shape under low-oxygen conditions. These distorted cells lose their flexibility and can obstruct small blood vessels.
However, modern research shows that the disorder extends far beyond reduced hemoglobin levels. It is now recognized as a systemic vascular and inflammatory condition that affects multiple organs over a lifetime2.
Although the disease originates from a single genetic mutation in hemoglobin, its downstream effects are very complex. Abnormally shaped red blood cells disrupt circulation, damage vascular tissues, and trigger chronic inflammatory responses3. As a result, patients experience recurring pain crises, progressive organ injury, and increased susceptibility to infection.
Earlier treatment strategies focused largely on correcting anemia and managing childhood complications4. Today, the understanding of SCD has expanded significantly. Researchers now view the disease as a dynamic interaction between abnormal red blood cells, vascular endothelium, and immune signaling pathways. This shift has transformed treatment objectives, placing greater emphasis on disease modification and, progressively, the pursuit of cure5.
Epidemiology and the malaria selection hypothesis
Because the mutation has persisted through evolutionary selection, sickle cell disease is now one of the most common inherited blood disorders worldwide. The highest prevalence occurs in sub-Saharan Africa, the Middle East, India, and parts of the Mediterranean6. Migration and population movement have gradually expanded the disease into North America and Europe7.
The geographic distribution reflects a well-known evolutionary phenomenon known as the malaria selection hypothesis8. Individuals who carry one copy of the sickle mutation, referred to as sickle cell trait, show partial protection against severe malaria infection. Because this survival advantage protected carriers from severe malaria, the mutation persisted in regions where the infection was historically endemic.
However, when a child inherits the mutated gene from both parents, the protective benefit disappears. Instead, the individual develops SCD, which introduces lifelong medical complications.
The shift from pediatric management to lifelong care
Advances in pediatric care have dramatically improved survival among children with SCD. Newborn screening programs, vaccination strategies, and early antibiotic prophylaxis have significantly reduced childhood mortality9-11.
As a result, many patients now survive well into adulthood. This demographic shift has changed the clinical focus of SCD management. Coordinated care across multiple medical specialties is now required to address chronic complications affecting the kidneys, lungs, brain, and cardiovascular system. Preventive strategies, early detection of organ damage, and ongoing monitoring have therefore become central components of modern sickle cell care1, 12, 13.
The molecular basis of sickle cell disease
The clinical features of sickle cell disease ultimately arise from a single genetic alteration in hemoglobin that changes the physical behavior of red blood cells.
Point mutation in β-globin
Sickle cell disease originates from a single point mutation in the β-globin gene located on chromosome 1114. This mutation replaces the amino acid glutamic acid with valine at the 6th position of the β-globin chain. Although the change appears small, it significantly alters the physical behavior of the hemoglobin molecule.
The resulting variant, HbS, functions normally when oxygen levels are high. However, under low-oxygen conditions the molecules interact abnormally and begin to aggregate. These abnormal molecular interactions initiate the structural processes that ultimately distort red blood cells into the sickle shape characteristic of SCD.
Molecular pathophysiology
Hemoglobin S polymerization
The defining molecular event in SCD is the polymerization of HbS during deoxygenation15. Instead of remaining dissolved within the red blood cell, HbS molecules assemble into long, rigid fibers. These fibers push against the cell membrane, distorting the normally flexible red blood cell into an elongated, crescent-shaped structure. Because these sickled cells are rigid, they struggle to pass through small capillaries.
Repeated cycles of oxygenation and deoxygenation worsen this process. Each cycle promotes additional polymer formation, which progressively damages the red cell membrane. Over time, many cells lose their flexibility permanently, further compromising blood flow through the microvasculature.
The erythrocyte lifecycle
Healthy red blood cells typically survive in circulation for about 120 days. In SCD, repeated membrane damage dramatically shortens this lifespan. Sickled erythrocytes become fragile and prone to rupture within the bloodstream, a process known as hemolysis, and their survival often falls to approximately 10–20 days16.
Although the bone marrow attempts to compensate by increasing red blood cell production, the rate of destruction frequently exceeds production. This imbalance produces chronic hemolytic anemia and contributes to systemic complications throughout the body17.
The sticky endothelium and flow obstruction
Abnormal red blood cell shape alone does not fully explain sickle cell complications. The vascular environment itself becomes highly adhesive during the disease process18.
Sickled erythrocytes display altered surface molecules that increase their attachment to endothelial cells lining blood vessels. At the same time, inflammatory signals activate the endothelium, which further promotes cellular adhesion18. Leukocytes and platelets also participate in these interactions, creating clusters of cells within small blood vessels. As these aggregates accumulate, blood flow slows progressively and microvascular obstruction develops. When this occurs, tissues experience reduced oxygen delivery, which triggers ischemic injury and severe pain.
The pathophysiological cascade
Sickle cell disease produces a complex pathophysiological cascade that extends beyond abnormal red blood cell shape. Repeated cycles of vascular obstruction, hemolysis, and inflammatory activation gradually transform a localized hematologic defect into a systemic vascular disorder19.
Acute vaso-occlusive crises
Vaso-occlusive crises represent the most recognizable clinical feature of SCD20. During these episodes, aggregates of sickled cells obstruct small blood vessels and interrupt normal blood flow. Restricted circulation deprives tissues of oxygen, which produces ischemic injury and intense pain. Nerve endings within affected tissues respond to this damage by generating strong pain signals.
Various factors can trigger these episodes, including dehydration, infection, temperature changes, and physiological stress. Regardless of the trigger, the underlying mechanism remains impaired microvascular circulation caused by sickled red blood cells and vascular adhesion.
Chronic endothelial dysfunction and inflammation
In addition to acute crises, SCD produces persistent vascular inflammation21. Repeated episodes of vaso-occlusion damage endothelial cells lining the blood vessels. This injury stimulates inflammatory signaling pathways that recruit immune cells and amplify vascular activation. Cytokine release further increases adhesion molecule expression on endothelial surfaces. As a result, blood cells attach more easily to vessel walls, which reinforces the cycle of microvascular occlusion and inflammation.
Hemolysis and its systemic fallout
In addition to vascular obstruction, chronic hemolysis represents another major driver of systemic complications in SCD22. When fragile sickled erythrocytes rupture within the circulation, intracellular components are released into the bloodstream, which disrupts vascular signaling and promotes oxidative injury.
Nitric oxide scavenging
Hemolysis releases free hemoglobin into the bloodstream. This molecule binds nitric oxide, which normally promotes vasodilation and regulates vascular tone23. As nitric oxide availability decreases, blood vessels lose their ability to relax efficiently. The resulting vascular dysfunction contributes to complications such as pulmonary hypertension and endothelial injury24.
Free heme and oxidative stress
In addition to free hemoglobin, hemolysis releases heme molecules that activate inflammatory pathways. Free heme stimulates oxidative reactions and generates reactive oxygen species25. These molecules damage vascular structures and cellular membranes. Over time, oxidative injury contributes to chronic organ dysfunction throughout the body.
Clinical manifestations
The acute presentation
Acute complications represent the most frequent cause of hospitalization in sickle cell disease. These events arise primarily from microvascular occlusion and organ ischemia, which produce several characteristic clinical syndromes26, 27.
Vaso-occlusive crisis and acute chest syndrome
Vaso-occlusive pain crises represent the most common acute manifestation of SCD, typically affecting bones, joints, and the chest. Acute chest syndrome is a particularly serious complication20. It occurs when sickled cells obstruct pulmonary vessels, which produces inflammation within lung tissue. Patients often develop chest pain, fever, and respiratory distress, and the condition may progress rapidly.
Stroke and neurovascular complications
In addition to pain crises, SCD also produces serious neurological complications28. Children with SCD face a markedly increased risk of stroke due to abnormal cerebral blood flow. Narrowed arteries and vascular injury contribute to this vulnerability. Silent cerebral infarctions may also occur without obvious symptoms, yet they can impair cognitive development and academic performance.
Splenic sequestration and infections
Another important complication involves the spleen29. Repeated vaso-occlusion damages the spleen over time. As splenic function declines, the body loses an important defense against bacterial infections. For this reason, early vaccination and preventive antibiotic therapy remain essential components of pediatric care30.
Long-term organ remodeling
Beyond acute complications, repeated vascular injury gradually produces long-term structural damage in multiple organs. Kidney damage may lead to proteinuria and progressive renal insufficiency31. Pulmonary hypertension increases strain on the right side of the heart and may eventually cause cardiac dysfunction32.
Bone tissue is also vulnerable to impaired circulation. Reduced blood supply can produce avascular necrosis, particularly in the hip and shoulder joints33. These complications highlight the systemic nature of the disease.
Precision phenotyping and biological diversity
Sickle cell disease shows remarkable clinical variability. The most severe form, HbSS disease, occurs when two sickle genes are inherited. Other variants, such as HbSC disease and HbS-β thalassemia, often produce different patterns of complications and disease severity34, 35.
In addition to these primary genetic combinations, several biological modifiers influence disease expression. One of the most important modifiers is fetal hemoglobin (HbF) 36. Because HbF does not participate in sickling polymer formation, higher HbF levels protect red blood cells from deformation.
Other genetic variants that regulate HbF production or inflammatory signaling pathways can further modify disease severity. This biological diversity explains why patients with the same primary mutation may experience very different clinical courses. Because of this, modern medicine increasingly relies on personalized analysis to tailor treatments to an individual’s unique genetic signature. This shift toward precision medicine helps explain why therapies designed for a single disease model have historically produced inconsistent clinical outcomes.
Managing and modifying the disease
New insights into sickle cell biology have led to major changes in treatment approaches. Today, care includes both traditional supportive measures and specific therapies that directly address the disease.
Traditional standard of care
Long-term treatment continues to rely primarily on hydroxyurea37. The drug increases fetal hemoglobin production, which interferes with HbS polymerization and reduces sickling events. Hydroxyurea also lowers leukocyte counts and inflammatory signaling, thereby decreasing vaso-occlusive crises.
In addition to pharmacologic therapy, blood transfusion strategies remain an important component of disease management38. Transfusions are often used to treat severe anemia and prevent stroke. By introducing healthy donor red blood cells, transfusions improve oxygen delivery and dilute sickled cells in circulation. However, repeated transfusions may lead to iron overload and alloimmunization, which requires careful monitoring39.
The era of disease-modifying agents
Newer therapies target specific biological pathways involved in sickle cell pathology. For example, voxelotor stabilizes hemoglobin in its oxygenated state, which reduces polymer formation and improves red cell survival40.
Other treatments focus on vascular adhesion rather than hemoglobin structure. Crizanlizumab blocks P-selectin, an adhesion molecule that promotes interactions between blood cells and the vascular endothelium41. By inhibiting this pathway, the therapy reduces the frequency of vaso-occlusive crises.
In contrast, L-glutamine therapy targets oxidative stress within red blood cells42. The treatment restores cellular antioxidant capacity, which improves membrane resilience and reduces oxidative injury.
The genomic revolution
Recent advances in genomic medicine have introduced new strategies aimed not only at controlling symptoms but at correcting the underlying genetic defect of sickle cell disease.
Allogeneic hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation currently represents the most established curative therapy within the realm of regenerative medicine for SCD43. The procedure replaces diseased bone marrow with healthy donor stem cells that produce normal hemoglobin. When successful, the transplant can eliminate sickling and restore normal red blood cell function. However, the approach is limited by the availability of compatible donors and the risks associated with transplantation.
The CRISPR/Cas9 breakthrough
Recent advances in gene editing have introduced alternative curative strategies that modify a patient’s own stem cells. Using CRISPR/Cas9 technology, researchers can alter genetic sequences responsible for regulating hemoglobin expression44. One successful approach targets the BCL11A gene, which suppresses fetal hemoglobin production after birth. By disrupting this regulatory pathway, gene editing reactivates HbF synthesis and prevents sickling.
Lentiviral vector gene therapy
A complementary strategy uses lentiviral vectors to introduce functional β-globin genes into hematopoietic stem cells45. These modified cells can then produce functional hemoglobin after transplantation back into the patient. Early clinical trials have produced encouraging results. However, the high cost and specialized infrastructure required for these therapies continue to limit their global accessibility.
Digital health and the future clinic
Advances in genomics are not the only forces reshaping sickle cell care. Emerging digital health technologies may also improve long-term disease management and monitoring.
Artificial intelligence models can analyze clinical data to identify patterns that precede vaso-occlusive crises, enabling earlier intervention46. Wearable monitoring devices can also provide continuous physiological data, including heart rate and oxygen saturation47. Together, these tools may help detect early signs of physiological stress before severe symptoms develop.
As survival improves, healthcare systems must also address the needs of an aging sickle cell population. Long-term monitoring for cardiovascular, renal, and neurological complications will become increasingly important.
Survivorship and the silent burden
Longer survival has revealed additional challenges that extend beyond acute medical complications. Repeated vascular injury can affect brain development and cognitive function. Even without overt stroke, subtle neurological damage may impair attention and memory48.
Chronic pain and frequent hospitalizations also create significant psychosocial stress. Patients often experience disruptions in education, employment, and social relationships49. Stigma related to opioid use for pain management may further complicate care50.
For these reasons, long-term management increasingly relies on multidisciplinary follow-up involving hematologists, neurologists, psychologists, and rehabilitation specialists.
From crisis management to precision recovery
The understanding of SCD has evolved from a simple description of inherited anemia to a complex model of vascular and inflammatory dysfunction. This shift has transformed both diagnosis and treatment strategies.
Modern therapies increasingly target the molecular drivers of the disease, while gene editing technologies bring the possibility of definitive cures closer to reality. However, ensuring equitable access to these innovations remains a major global challenge. Bridging the gap between scientific progress and healthcare delivery will be essential for improving outcomes worldwide.